- Review
- Open access
- Published:
Molecular genetics of microsatellite-unstable colorectal cancer for pathologists
Diagnostic Pathology volume 12, Article number: 24 (2017)
Abstract
Background
Microsatellite-unstable colorectal cancers (CRC) that are due to deficient DNA mismatch repair (dMMR) represent approximately 15% of all CRCs in the United States. These microsatellite-unstable CRCs represent a heterogenous group of diseases with distinct oncogenesis pathways. There are overlapping clinicopathologic features between some of these groups, but many important differences are present. Therefore, determination of the etiology for the dMMR is vital for proper patient management and follow-up.
Main body
Epigenetic inactivation of MLH1 MMR gene (sporadic microsatellite-unstable CRC) and germline mutation in an MMR gene (Lynch syndrome, LS) are the two most common mechanisms in the pathogenesis of microsatellite instability in CRC. However, in a subset of dMMR CRC cases that are identified by screening tests, no known LS-associated genetic alterations are appreciated by current genetic analysis. When the etiology for dMMR is unclear, it leads to patient anxiety and creates challenges for clinical management.
Conclusion
It is critical to distinguish LS patients from other patients with tumors due to dMMR, so that the proper screening protocol can be employed for the patients and their families, with the goal to save lives while avoiding unnecessary anxiety and costs. This review summarizes the major pathogenesis pathways of dMMR CRCs, their clinicopathologic features, and practical screening suggestions. In addition, we include frequently asked questions for MMR immunohistochemistry interpretation.
Background
Colorectal cancer (CRC) represents the third most common malignancy diagnosed both in men and women in the United States. Approximately 15% of CRC display a defect in the mismatch repair pathway [1], resulting in microsatellite instability (MSI). The identification of MSI CRC from microsatellite-stable (MSS) tumors is clinically important, because MSI tumors have a better stage-adjusted survival compared to MSS tumors and may respond differently to 5-fluorouracil-based adjuvant chemotherapy [2]. In addition, it is important to identify those patients with Lynch Syndrome (LS).
Defects in the mismatch repair (dMMR) mechanism leads to the pathogenesis of MSI tumors. The MMR mechanism identifies and fixes base-pair mismatches that occur within the genome. Proteins within the MMR system include MLH1, PMS2, MSH2, MSH6, MLH3, MSH3, PMS1, and Exo1. These proteins form heterodimers that repair DNA damage. The most common and relevant heterodimers in colorectal carcinogenesis are MLH1/PMS2 and MSH2/MSH6. Microsatellites, also known as short tandem repeats, are composed of mono- to hexa-nucleotides that constitute a repeated motif. These microsatellites constitute up to 3% of the genome and are variable in length. Chromosomal alleles often contain different lengths of the same microsatellite. Due to their repetitive nature, microsatellites are highly susceptible to errors in MMR; therefore dMMR tumors demonstrate microsatellite instability and are often hyper- or ultra-mutated [3].
MSI CRC identified by screening tests (MMR immunohistochemistry, IHC, or polymerase chain reaction, PCR) can be further divided into four categories after additional testing [4] (also see Table 1):
-
(A)
Sporadic dMMR – MLH1 promoter hypermethylation: MSI CRC due to hypermethylation of CpG islands in the MLH1 promoter (these tumors often arise via the serrated pathway, harbor BRAF mutation, and account for 10% to 15% of CRC);
-
(B)
Lynch Syndrome due to germline mutations in one of the MMR genes (MLH1, PMS2, MSH2, MSH6) or alteration in EPCAM (TACSTD1) gene that causes epigenetic silencing of MSH2 [5] (these tumors often arise in tubular adenomas and account for approximately 3% of CRC);
-
(C)
Unexplained dMMR Colorectal Cancers (sporadic dMMR-somatic MMR mutation and others): Cases with neither identified germline mutation of MMR nor hypermethylation of the promoter region of MLH1 (these cases have unexplained dMMR and have been termed Lynch-like by some);
-
(D)
Rarely, constitutional MMR deficiency syndrome (biallelic germline MMR mutations; of note, the normal adjacent tissue in these cases will also have dMMR).
Main text
Sporadic dMMR colorectal cancers – MLH1 promoter hypermethylation/CpG Island Methylator Phenotype (CIMP)
Tumors that show DNA hypermethylation at multiple promoters are classified as showing the CpG Island Methylator Phenotype (CIMP). There are several types of epigenetic alterations, including DNA hypermethylation, that regulate gene expression. Promoter methylation suppresses gene transcription by inhibiting binding of transcription factors, affecting histone acetylation, and altering conformations to effectively block access of transcriptional machinery to the gene. Epigenetic silencing in tumors is biologically equivalent to acquiring an inactivating mutation and may account for the first, second, or both hits in silencing tumor suppressor genes. Genes that are commonly hypermethylated in CRC include MLH1, MCC (methylated in CRC), APC and MGMT. The promoter region of five genes has been chosen as proxy markers of CIMP: CACNA1G, IGF2, NEUROG1, RUNX3 and SOCS1 [6]. Hypermethylation of at least three markers is the definition of CIMP. CRC with CIMP frequently harbor BRAF mutations and show methylation of MLH1 resulting in MSI in up to 70% of cases. Of interest, the CDX2 negative/Cytokeratin 20 negative immunohistochemical phenotype defines a distinct subgroup of MSI CRCs with poor differentiation, CIMP status, and unfavorable prognosis [7].
Lynch syndrome
LS is an autosomal dominant disorder that increases the risk of developing CRC and endometrial adenocarcinoma, as well as tumors of the small intestine, stomach, ureter, renal pelvis, ovary, brain, prostate [8], among others. LS is a result of deleterious germline mutations in the genes associated with DNA MMR (MLH1, PMS2, MSH2, MSH6 and EPCAM). Cancers (such as CRC) arise when a second hit occurs in the unaffected wild type allele, through various mechanisms such as loss of heterozygosity, mutation, or hypermethylation. Most (90%) CRC due to LS have MSI due to the defective MMR mechanisms caused by the germline mutation in association with the second hit. It is the most common hereditary CRC syndrome, with approximately 2–5% of all CRCs due to LS. Patients with LS benefit from increased surveillance; therefore, identification of patients as well as family members with this syndrome is very important. Historically, these cases were grouped with other familial CRC cases and referred to as Hereditary Nonpolyposis Colorectal Cancer (HNPCC). The term HNPCC is no longer preferred due to observations that patients with LS do have adenomatous polyps and these adenomatous polyps are more likely to progress to CRC [9].
The Amsterdam criteria were initially developed in 1990 [10] and later revised to Amsterdam II criteria in 1998 [11], to help identify families likely to have HNPCC for research purposes by using personal and family histories. The Amsterdam criteria include a series of clinical criteria that are also known as the “3-2-1” rule (Table 2). The Bethesda guidelines were developed in 1997 to identify patients who should have tumor screening for LS [12]. The revised Bethesda guidelines proposed in 2004 [13] incorporated histologic components into the screening guidelines. A comparison of the Amsterdam criteria and the revised Bethesda criteria is detailed in Table 2. Unfortunately, a significant portion of CRC patients with LS were still missed when using these criteria alone without MMR IHC or PCR testing [14]. Therefore, it is currently recommended that all newly diagnosed patients with CRC undergo LS screening by IHC and/or MSI PCR testing as recommended by many authoritative organizations, including the Evaluation of Genomic Applications in Practice and Prevention (a working group sponsored by the Centers for Disease Control and Prevention) in 2009 [15], the National Comprehensive Cancer Network in 2014 [16], the US Multi-Society Task Force in 2014 [17], the American College of Gastroenterology [18] and the American Society of Clinical Oncology [19] in 2015. Patients who have clinical features concerning for LS, but whose screening results indicate microsatellite stability, should be further evaluated by another technique such as MSI PCR (if initial screening was by IHC staining) or IHC for the MMR proteins (if initial screening was by MSI molecular testing), or possibly genetic sequencing of MMR genes since the sensitivity of the MSI by PCR and IHC is not 100%.
Phenotypic variation in LS
The clinical presentation of a patient with LS can vary depending on the MMR gene affected in the germline. Patients with MLH1 mutations typically present with classic LS (CRC as the first presenting symptom with a mean age of 43 to 46). Similarly, patients with MSH2 mutations also present with classic LS, however these patients are also at increased risk of extra-colonic cancers. Muir-Torre syndrome, which is a rare variant of LS in which patients develop hair follicle and sebaceous gland neoplasms, have MSH2 germline mutations. Patients with germline mutations in MSH6 are more likely to develop endometrial cancer and may not test positive for MSI by PCR. Mutations in PMS2 tend to develop CRC at an older age compared to classic LS. The rare biallelic mutation in any of the four MMR genes (constitutional mismatch repair deficiency syndrome) manifests as very early-onset (pediatric) hematological, colorectal, urinary tract and brain (glioblastoma) cancers, and neurofibromatosis [9].
Unexplained dMMR colorectal cancers (sporadic dMMR-somatic MMR mutation and others)
This is an ill-defined group of MSI CRC patients who have discordant screening and germline results. In these cases, the initial screening for LS is positive (either abnormal MMR IHC or MSI PCR); however, BRAF V600E PCR and/or MLH1 promoter hypermethylation studies does not indicate sporadic CRC, and genetic sequencing for MMR genes do not identify a germline mutation. These unexplained dMMR cases have been referred to as “Lynch-like syndrome” in the literature, however the causes for these cases are heterogenous and Lynch-like is a misleading nomenclature.
Recent studies have elucidated some of the causes/errors that could make a tumor look like a sporadic dMMR tumor. Approximately 70% of these patients may have biallelic somatic mutations in the MMR gene [20–22]. These patients with biallelic somatic mutations do not carry a germline mutation in MMR genes and therefore their progeny are not at risk of LS, and do not need lifelong intense screening protocol like LS patients.
Other potential explanations for unexplained dMMR cases [23] include: (a) incorrect performance/interpretation of MMR IHC results (absence of nuclear staining due to technical limitations or professional misinterpretations) (20%); (b) failure to detect germline MMR gene alteration using currently available tests (such as inversion of MSH2 exons 1 to 7 that is not tested by many commercial labs) (unknown%); (c) other germline gene defects that also cause MSI phenotype, such as biallelic MUTYH mutation (3%) [24–26]; (d) somatic mosaicism (<1%) [27–29]; (e) constitutional epimutation of MLH1 (germline monoallelic hypermethylation of the MLH1 promoter region; no alteration to the genetic sequence of MLH1) (<1%) [30–32].
Constitutional mismatch repair deficiency syndrome (CMMRD)
CMMRD patients have germline mutation of both alleles of a MMR gene (MLH1, PMS2, MSH2, MSH6), leading to loss of expression of the corresponding MMR protein. Unlike LS, CMMRD patients show loss of MMR staining in both the tumor and the background non-neoplastic tissue.
Clinicopathologic features of MSI CRC
Clinically, tumors with MSI are more common in the right colon [33]. Sporadic tumors typically occur in older female patients, whereas, CRC in the context of LS often occurs in younger patients (50 years of age or less). These tumors respond poorly to 5-fluorouracil based chemotherapies [34–36], but patients with MSI tumors have a better prognosis than those with stage- matched MSS tumors [37–40].
Microscopically, MSI tumors share similar histomorphology regardless of their respective pathogenesis. They frequently have a mucinous phenotype, and are either very well-differentiated or poorly differentiated. The presence of signet ring cells or a medullary phenotype is also frequently seen in this context. Intratumoral heterogeneity (mixed conventional, mucinous, and poorly differentiated carcinoma) is common. The “dirty necrosis” seen in microsatellite-stable colon cancers is frequently absent. Tumors often contain increased intratumoral lymphocytes, and may have a Crohn’s -like reaction with prominent lymphoid aggregates at the periphery of the tumor [13, 41–43].
The histologic features of germline and sporadic MSI tumors are similar, although finding an adjacent sessile serrated adenoma as the precursor lesion is more suggestive of a sporadic tumor. In contrast, the precursor lesion associated with LS is a tubular adenoma. A recent study by Mas-Moya et al [44] demonstrated that when compared to LS, Lynch-like syndrome is more likely to occur in the right colon (a subset of LS tumors occurs at left colon [45]), more often identified in patients with tumors that harbor concurrent MLH1/PMS2 loss (with lack of MLH1 promoter hypermethylation) or concurrent MSH2/MSH6 loss, and less likely to have synchronous or metachronous LS-associated cancers.
The prognosis of patients with CRC associated with LS and MLH1 promoter hypermethylation appear to be similar [46]. In up to 70% of CRC with hypermethylation of the MLH1 promoter, there is a BRAF V600E gene mutation. CRC patients with BRAF V600E mutation have been shown to have a limited clinical response to epidermal growth factor receptor (EGFR) targeted therapies (cetuximab or panitumumab). Recent studies indicated that therapies targeting the PD-1 immune checkpoint blockade are effective in treating dMMR CRC [47].
Screening for MSI and LS
Screening for MMR function can be done using IHC for the MMR proteins, and/or MSI analysis by polymerase chain reaction (PCR).
MMR protein expression by IHC
is performed to detect the absence or loss of a particular protein within the nucleus of the tumor cells; abnormal IHC correlates strongly with MSI status by PCR. MMR proteins are normally present in many human cells, particularly in proliferating cells such as crypt epithelium. Understanding the biochemistry/molecular biology of MMR proteins is important in the interpretation of IHC results. The most common mutations found in LS are frameshift or nonsense mutations that cause protein truncation or increased degradation. These mutations result in absence/loss of the mutated protein, which is easily detected by IHC. The MMR proteins MLH1, PMS2, MSH2 and MSH6 form heterodimer pairs in vivo (MLH1/PMS2 (MutLalpha) and MSH2/MSH6 (MutSalpha)). When either MLH1 or MSH2 is mutated and degraded in vivo, this will cause subsequent degradation and protein loss of its partner PMS2 or MSH6, respectively. For instance, when MSH2 is mutated, this will appear immunohistochemically as absence/loss of MSH2 and MSH6. However, the opposite situation is not typically found. When either germline PMS2 or MSH6 is mutated and therefore degraded in vivo, there is usually no concomitant loss of protein expression of MLH1 or MSH2, respectively. This is due to the compensatory effects of other MMR proteins such as MSH3, which can bind to, and stabilize, either MLH1 or MSH2. Tumors that show absence of MSH2 and/or MSH6 or PMS2 are suspicious for LS, and these patients should be considered for sequencing of whatever protein was missing (after proper consent). If a germline mutation is then identified, this is diagnostic of LS. If a germline mutation is not identified, somatic MMR gene and/or loss of heterozygosity testing may be considered.
MSI by PCR
evaluates tumors by amplifying microsatellite repeats. The most widely used panel, the Bethesda panel, consists of five microsatellite repeats, including 2 mononucleotide repeats (BAT25 and BAT26) and 3 dinucleotide repeats (D2S123, D5S346, and D17S250) [48]. Microsatellites demonstrating at least two novel alleles (shift) define MSI high (considered to be microsatellite unstable). MSI low is defined as the detection of only one unique (shifted) microsatellite, although the significance of this category is controversial. When no shifted microsatellites are identified, this is defined as MSS. If MSI analysis is used as the initial screening test rather than IHC, BRAF mutational testing or MLH1 promoter methylation can be used as the second step prior to genetic sequencing in patients found to have MSI.
Either MMR IHC or MSI by PCR works well for screening most cases, but each has limitations. IHC labs are much more widely available than molecular labs, but IHC results may be more affected by tissue fixation conditions. MSI analysis requires normal tissue in addition to tumor tissue for comparison, and may require tumor microdissection, but is not typically affected by fixation. Individual laboratories and multidisciplinary teams ultimately must determine for themselves which technique(s) is most effective in their institution. This issue may become less important with the advent of next generation sequencing panels [49], in which the cost for testing for all the MMR genes may in some laboratories be similar to testing for a one single MMR gene. Of note, MSI by PCR and MMR by IHC rarely may miss MSH6 mutations, because MSH6 mutations are often MSI-low or MSS. This is a known pitfall in MSI testing for gynecologic cancers given a larger proportion of endometrial cancers harbor MSH6 mutations compared to CRC.
Approximately 5 to 10% of all CRC have substitution of valine with glutamic acid at amino acid position 600 of BRAF. BRAF is a protein in the mitogen active protein kinase pathway and acts as a serine/threonine kinase. The BRAF V600E mutation causes constitutive activation of the kinase and increases MAP kinase signaling. CRC with BRAF mutations are more commonly found in the right colon, have a high histologic grade, show female predominance, and more frequently originate from serrated polyps. Importantly, BRAF V600E mutations are almost never found in LS, but are seen in 40–76% of sporadic MSI CRCs. Therefore, in screening algorithms for patients with CRC, analysis of BRAF is a cost-effective method to screen for patients who should undergo further genetic testing for LS rather than gene sequencing in all those with absence of MLH1 and PMS2 [50, 51].
Besides being useful in distinguishing sporadic CRCs from LS, BRAF V600E also has prognostic value, as mutations have been suggested to be associated with a worse outcome in MSS CRC. It may also predict resistance to EGFR mediated antibodies [52]. Since BRAF and KRAS are part of the same MAP kinase pathway, it is not surprising that BRAF V600E mutations are almost mutually exclusive of KRAS mutations. Of note, BRAF mutation testing has no role in many extracolonic LS-associated cancers, including endometrial and ovarian cancers [53].
In patients with MLH1-deficient CRC, direct testing of MLH1 promoter hypermethylation and/or BRAF V600E mutational analysis, may be a cost-effective means of identifying patients with sporadic tumors for whom germline genetic testing is not indicated.
Figure 1 demonstrates the algorithm we currently use for screening newly diagnosed CRC for LS. Initially, the cancer is immunostained for MLH1, MSH2, MSH6, and PMS2. When all four proteins are present, no further work up is necessary unless there is specific clinical concern. When MLH1 and PMS2 are absent by IHC, the next step is analysis of BRAF by either PCR or IHC [50, 51, 54, 55] to help determine if the patient has a sporadic tumor or should be further evaluated for LS. We use PCR rather than IHC for BRAF. If BRAF V600E mutation is found, no further testing is required, as the tumor is presumed to be sporadic CRC. If no BRAF mutation is detected, the subsequent step depends on the degree of suspicion of LS based on the patient’s age and history. If there is suspicion of LS, the tumor will be sent for genetic sequencing of MLH1 or PMS2. Conversely, if there is low suspicion of LS, MLH1 promoter hypermethylation is then analyzed. If MLH1 promoter hypermethylation is not detected, the patient will be sent for sequencing of MLH1 or PMS2. Some laboratories use MLH1 methylation testing rather than BRAF mutational analysis after IHC in those tumors found to have absence of MLH1 and PMS2. Similar to BRAF mutated cases, those with MLH1 methylation are presumed to represent sporadic tumors and do not need to undergo further molecular testing for LS.

Algorithm for screening newly diagnosed colorectal carcinoma (CRC) for Lynch syndrome (LS). The initial step is to screen cases using immunohistochemistry (IHC) for mismatch repair proteins. When MLH1 and PMS2 are absent, the subsequent step is to utilize BRAF analysis by polymerase chain reaction (PCR) or immunohistochemistry
Practical guide for MMR IHC interpretation
MMR sensitivity and specificity
MMR IHC has good sensitivity for MSI-high tumors (>90%) [56–59]; however it suffers lower sensitivity for MSI-low tumors. In terms of specificity, MMR IHC is efficient in detecting LS with MSH2 and MSH6 mutation; however specificity for MLH1 and PMS2 is low due to MLH1 hypermethylation [60, 61]. MLH1 and PMS2 often show loss of expression in sporadic CRC due to transcriptional silencing of MLH1 by promoter hypermethylation.
MMR staining patterns
Most cases screened with MMR IHC show typical patterns of staining and the interpretation is relatively straight forward. MMR proteins function in the cell nuclei, therefore the staining pattern is nuclear. However, punctate/speckled nuclear staining and nuclear membrane staining are considered abnormal staining patterns that should not be interpreted as preserved MMR protein expression [23]. They either represent loss of expression or staining artifact and therefore warrant additional investigation (compare staining to the internal control, repeat on a different block or sample, or MSI by PCR).
It is important to point out that it is common to see patchy (intratumoral heterogeneity) MMR staining in MSS tumors. Tissue hypoxia and fixation issues have been implicated for the staining heterogeneity [62, 63]. No definite consensus is present regarding the minimal percentage of positive nuclei to be present to indicate preserved/intact expression. In our practice, if more than 5% tumor nuclei show unequivocal nuclear staining (some use >10% [23], or any convincing staining [64]), it is considered normal/intact staining pattern. However, if less than but close to 5% tumor nuclei are positive, repeating the stain is the next step. If the repeat is similar, the stain result is considered equivocal and additional testing (MSI by PCR or genetic) is suggested. Of note, one needs to always make sure the positive internal control (stromal cells, lymphocytes, or nonneoplastic epithelial cells), shows nuclear positivity, before calling ‘loss of staining’ in the tumor cells.
It is advisable not to use ‘positive’ or ‘negative’ when reporting MMR IHC results. In the case of MMR IHC, positive (presence of) staining is a normal result, while negative (lack of) staining is abnormal and suggests the need for further testing. Therefore, the conventional “positive” and “negative” descriptors can be confusing and should be avoided in MMR IHC reports. We prefer to use the terminology “intact” when tumor nuclei show staining, and “loss” when tumor nuclei lack staining and this language is used in the College of American Pathologists (CAP) biomarker protocol [64].
As discussed previously, the pairing and dimerization of the MMR proteins stabilize the function unit, which help to explain the staining pattern of MMR expression/loss observed. Figures 2 and 3 demonstrate two cases with abnormal MMR IHC results. Figure 2 demonstrates MMR IHC in a patient with sporadic hypermethylation of the MLH1 promoter, in which both MLH1 and PMS2 show loss of expression. Figure 3 demonstrates MMR IHC in a patient with LS with isolated loss of MSH6.

Immunohistochemistry for mismatch repair proteins in a patient with sporadic hypermethylation of the MLH1 promoter. Hematoxylin & eosin (H&E) stain of the adenocarcinoma (a). Tumor cells show loss of MLH1 nuclear expression (b) and loss of PMS2 nuclear expression (c) while the stroma and lymphocytes shows strong intact staining. Conversely, tumor cells show intact nuclear expression of MSH2 (d) and MSH6 (e). This tumor showed BRAF V600E mutation by PCR consistent with sporadic microsatellite-unstable colorectal carcinoma

Immunohistochemistry for mismatch repair proteins in a patient with Lynch syndrome. H&E stain of the mucinous adenocarcinoma (a). Intact nuclear expression of MLH1 (b) and PMS2 (c). Intact nuclear expression of MSH2 (d). This tumor showed absent nuclear staining of MSH6 (e). Genetic sequencing confirmed mutation of the MSH6 gene
When all four MMR proteins are absent, the possibility of poor fixation and loss of antigenicity should be considered. The surrounding stroma is an excellent positive control and should always show staining. On the other hand, intact expression of all four proteins does not exclude LS. About 5% of families harbor a missense mutation (most commonly in MLH1) that results in a nonfunctional protein with retained antigenicity.
Unusual Patterns That Can Lead to Confusion
-
In cases of isolated loss of PMS2 IHC, germline MLH1 analysis should be performed if no mutations are detected through PMS2 testing. In a study by Dudley et al [65], approximately 24% of patient with tumors with isolated loss of PMS2 expression harbor germline MLH1 mutation. Furthermore, a subset of MLH1 mutations result in functionally inactive MLH1 protein that is antigenically intact and will be detected by the commonly used anti-MLH1 antibody clones. Such germline MLH1 mutations will lead to decreased MLH1 protein stability and/or quantity, compromised stability of MLH1-PMS2 complexes, and subsequent PMS2 degradation. Recently, Rosty et al. [66] demonstrated similar findings, and they also recommend germline MLH1 mutation analysis in individuals with isolated loss of PMS2 expression but without PMS2 mutation identified.
-
Very rarely, all four stains are lost (null pattern) due to a germline MSH2 mutation (causing absent MSH2/MSH6) together with a somatic MLH1 hypermethylation (causing absent MLH1/PMS2) [67].
-
Another uncommon finding is loss of MSH6 together with MLH1 and PMS2. This could occur due to MLH1 promoter hypermethylation causing MLH1/PMS2 loss, leading to MSI. Such MLH1/PMS2 deficient, MSI tumors, are prone to generate somatic mutations in MSH6 gene, which cause significantly reduced staining of MSH6 [68].
FAQs for MMR immunohistochemistry
-
1.
A patient’s colon cancer was previously screened for LS and no abnormality was found, should I screen a second colon or endometrial cancer in this patient?
Answer: MMR IHC of synchronous/metachronous neoplasms in LS patients demonstrated discordant MMR immunoreactivity in 31% cases [69], therefore it may be worthwhile to perform LS screening in all primary, synchronous, and metachronous LS-associated neoplasms if a previous tumor screened intact.
-
2.
A Gastroenterologist asked to test MMR IHC in an adenoma, how should I proceed for the IHC interpretation?
Answer: The criteria for interpreting MMR IHC in an adenoma are the same as for cancer. It has been shown that 50% to 72% of conventional adenomas from mutation carriers show loss of MMR protein expression concordant with the underlying germline mutation [70, 71]. In addition, the absence of staining was particularly frequent in larger adenomas in LS patients, was associated with adenomas with villous component, and was observed more often in adenoma with high-grade dysplasia. However, some adenomas in LS patients only contain loss of one allele and do not contain the second hit; and therefore still demonstrate preserved MMR protein expression. In signing out these cases we comment that “whereas absent staining may be seen in LS and the pattern of loss useful in directing gene testing, intact expression does not exclude the diagnosis.”
-
3.
Does neoadjuvant therapy in rectal cancer affect MMR IHC result?
Answer: Rectal tumors treated with neoadjuvant therapy can sometimes show decreased or complete loss of MSH6 staining or only a nucleolar staining pattern (Fig. 4) [72, 73]. Evidence has shown that most of these cases do not have a MSH6 mutation and this should not be interpreted as loss/absence of MSH6. Rather than immediately sequencing questionable cases, repeating the IHC or performing IHC on the pretreatment biopsy will help resolve this issue. Decreased expression of PMS2 has also been reported in 30% of posttreatment rectal carcinomas [74]
Fig. 4 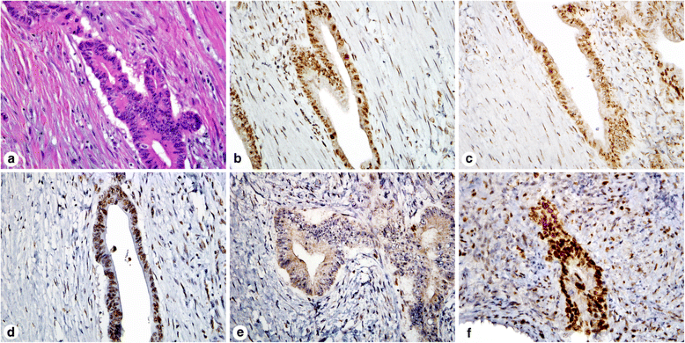
Immunohistochemistry for mismatch repair proteins in a patient that received neoadjuvant chemotherapy for rectal adenocarcinoma. H&E stain of the tumor in the resection specimen (a). The resection specimen showed intact MLH1 (b), PMS2 (c), and MSH2 (d) staining. MSH6 staining of the resection specimen showed focal nucleolar staining (e) that was originally interpreted as absent, but subsequent molecular sequencing did not reveal a mutation. The pretreatment tumor biopsy was stained for MSH6 and showed intact staining (f)
-
4.
Can MMR IHC be performed on autopsy cases?
Answer: Yes, MMR IHC may be performed on autopsy cases, but be aware of the inferior preservation of the tumor tissue in autopsy cases, which may lead to weak and patchy nuclear staining of the tumor cells. Nonetheless, if the internal control (adjacent benign epithelium, stroma or background lymphocytes) demonstrates similar weak staining, it is most likely that the MMR protein expression is intact/normal.
-
5.
Can MMR IHC be performed on metastasis?
Answer: Our group recently demonstrated [75] that in 50 primary CRC with metastasis, there is 100% concordance of MMR IHC results between primary and metastasis, suggesting that using metastatic tissue to identify patients with dMMR tumors (screen for LS or to aid in therapeutic choices) is feasible when the primary tissue is not available for testing.
-
6.
Should MMR IHC be performed on serrated polyps to help exclude LS?
Answer: No. CRC arising from the serrated pathway has different underlying molecular genetics than CRC arising from LS [76], although both may show dMMR. The genetic signature of CRC arising from serrated polyps often contains BRAF mutation and MLH1 promoter hypermethylation. In contrast, LS almost never harbors BRAF mutation.

Conclusions
Detecting LS is desirable because intensified clinical cancer surveillance saves lives. Identification of MSI CRC is important, as dMMR may serve as a screening tool for detecting LS, a prognostic marker for patient outcome, and a predictive marker for response to chemotherapy. Tumor DNA sequencing should be undertaken in unsolved cases of abnormal LS screening without identifiable germline mutation, to evaluate for somatic biallelic mutations, as it can explain two thirds of these cases and help guide genetic counseling and reduce patient anxiety. Next generation sequencing methodology has been employed for detection of molecular alterations in dMMR CRC and may replace the current method of detecting MSI by PCR techniques in the near future.
Abbreviations
- CIMP:
-
CpG Island Methylator Phenotype
- dMMR:
-
Deficient DNA mismatch repair
- HNPCC:
-
Hereditary Nonpolyposis Colorectal Cancer
- IHC:
-
Immunohistochemistry
- LS:
-
Lynch syndrome
- MMR:
-
Mismatch repair
- MSI:
-
Microsatellite instability
- MSS:
-
Microsatellite stability
- PCR:
-
Polymerase chain reaction
References
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363(6429):558–61.
Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. 2015;16(7):30.
Bellizzi AM, Frankel WL. Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv Anat Pathol. 2009;16(6):405–17.
Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J Gastroenterol. 2015;21(31):9253–61.
Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41(1):112–7.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–93.
Kim JH, Rhee YY, Bae JM, Cho NY, Kang GH. Loss of CDX2/CK20 expression is associated with poorly differentiated carcinoma, the CpG island methylator phenotype, and adverse prognosis in microsatellite-unstable colorectal cancer. Am J Surg Pathol. 2013;37(10):1532–41.
Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, de la Chapelle A, Goldberg RM. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16(7):553–7.
Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer. 2015;15(3):181–94.
Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34(5):424–5.
Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–6.
Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89(23):1758–62.
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–8.
Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, Hopper JL, Le Marchand L, Gallinger S, Newcomb PA, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308(15):1555–65.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41.
Hampel H. NCCN increases the emphasis on genetic/familial high-risk assessment in colorectal cancer. J Natl Compr Canc Netw. 2014;12(5 Suppl):829–31.
Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology. 2014;147(2):502–26.
Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–62. quiz 263.
Stoffel EM, Mangu PB, Gruber SB, Hamilton SR, Kalady MF, Lau MW, Lu KH, Roach N, Limburg PJ. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. 2015;33(2):209–17.
Geurts-Giele WR, Leenen CH, Dubbink HJ, Meijssen IC, Post E, Sleddens HF, Kuipers EJ, Goverde A, van den Ouweland AM, van Lier MG, et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol. 2014;234(4):548–59.
Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, Pritchard CC. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308–16. e1301.
Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, Goossens M, Ouchene H, Hendriks-Cornelissen SJ, Kwint MP, Hoogerbrugge N, Nagtegaal ID, Ligtenberg MJ. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146(3):643–6. e648.
Pai RK, Pai RK. A Practical Approach to the Evaluation of Gastrointestinal Tract Carcinomas for Lynch Syndrome. Am J Surg Pathol. 2016;40(4):e17–34.
Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet. 2002;30(2):227-232
Nielsen M, de Miranda NF, van Puijenbroek M, Jordanova ES, Middeldorp A, van Wezel T, van Eijk R, Tops CM, Vasen HF, Hes FJ, et al. Colorectal carcinomas in MUTYH-associated polyposis display histopathological similarities to microsatellite unstable carcinomas. BMC Cancer. 2009;9:184.
Morak M, Heidenreich B, Keller G, Hampel H, Laner A, de la Chapelle A, Holinski-Feder E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet. 2014;22(11):1334–7.
Buchanan DD, Rosty C, Clendenning M, Spurdle AB, Win AK. Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome). Appl Clin Genet. 2014;7:183–93.
Pastrello C, Fornasarig M, Pin E, Berto E, Pivetta B, Viel A. Somatic mosaicism in a patient with Lynch syndrome. Am J Med Genet A. 2009;149A(2):212–5.
Sourrouille I, Coulet F, Lefevre JH, Colas C, Eyries M, Svrcek M, Bardier-Dupas A, Parc Y, Soubrier F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer. 2013;12(1):27–33.
Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013;15(1):25–35.
Hitchins M, Williams R, Cheong K, Halani N, Lin VA, Packham D, Ku S, Buckle A, Hawkins N, Burn J, et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129(5):1392–9.
Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, Polly P, Goldblatt J, Ward RL. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5′UTR. Cancer Cell. 2011;20(2):200–13.
Lynch PM, Lynch HT, Harris RE. Hereditary proximal colonic cancer. Dis Colon Rectum. 1977;20(8):661–8.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–57.
Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28(20):3219–26.
Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126(2):394–401.
Lanza G, Gafa R, Santini A, Maestri I, Guerzoni L, Cavazzini L. Immunohistochemical test for MLH1 and MSH2 expression predicts clinical outcome in stage II and III colorectal cancer patients. J Clin Oncol. 2006;24(15):2359–67.
Samowitz WS, Curtin K, Ma KN, Schaffer D, Coleman LW, Leppert M, Slattery ML. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol Biomarkers Prev. 2001;10(9):917–23.
Sinicrope FA, Rego RL, Halling KC, Foster N, Sargent DJ, La Plant B, French AJ, Laurie JA, Goldberg RM, Thibodeau SN, et al. Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology. 2006;131(3):729–37.
Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012;18(6):1506–12.
Shia J, Holck S, Depetris G, Greenson JK, Klimstra DS. Lynch syndrome-associated neoplasms: a discussion on histopathology and immunohistochemistry. Fam Cancer. 2013;12(2):241–60.
Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91(12):2417–22.
Jass JR, Smyrk TC, Stewart SM, Lane MR, Lanspa SJ, Lynch HT. Pathology of hereditary non-polyposis colorectal cancer. Anticancer Res. 1994;14(4B):1631–4.
Mas-Moya J, Dudley B, Brand RE, Thull D, Bahary N, Nikiforova MN, Pai RK. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum Pathol. 2015;46(11):1616–25.
Hartman DJ, Brand RE, Hu H, Bahary N, Dudley B, Chiosea SI, Nikiforova MN, Pai RK. Lynch syndrome-associated colorectal carcinoma: frequent involvement of the left colon and rectum and late-onset presentation supports a universal screening approach. Hum Pathol. 2013;44(11):2518–28.
Haraldsdottir S, Hampel H, Wu C, Weng DY, Shields PG, Frankel WL, Pan X, de la Chapelle A, Goldberg RM, Bekaii-Saab T. Patients with colorectal cancer associated with Lynch syndrome and MLH1 promoter hypermethylation have similar prognoses. Genet Med. 2016.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20.
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57.
Pritchard CC, Smith C, Salipante SJ, Lee MK, Thornton AM, Nord AS, Gulden C, Kupfer SS, Swisher EM, Bennett RL, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012;14(4):357–66.
Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, Westra J, Frebourg T, Espin E, Armengol M, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41(9):664–8.
Jin M, Hampel H, Zhou X, Schunemann L, Yearsley M, Frankel WL. BRAF V600E mutation analysis simplifies the testing algorithm for Lynch syndrome. Am J Clin Pathol. 2013;140(2):177–83.
Wong R, Cunningham D. Using predictive biomarkers to select patients with advanced colorectal cancer for treatment with epidermal growth factor receptor antibodies. J Clin Oncol. 2008;26(35):5668–70.
Mills AM, Longacre TA. Lynch Syndrome Screening in the Gynecologic Tract: Current State of the Art. Am J Surg Pathol. 2016;40(4):e35–44.
Roth RM, Hampel H, Arnold CA, Yearsley MM, Marsh WL, Frankel WL. A modified Lynch syndrome screening algorithm in colon cancer: BRAF immunohistochemistry is efficacious and cost beneficial. Am J Clin Pathol. 2015;143(3):336–43.
Bellizzi AM. Screening for Lynch syndrome: a no-brainer: BRAF V600E mutation-specific immunohistochemistry: caveat emptor. Am J Clin Pathol. 2015;143(3):320–4.
Lindor NM, Burgart LJ, Leontovich O, Goldberg RM, Cunningham JM, Sargent DJ, Walsh-Vockley C, Petersen GM, Walsh MD, Leggett BA, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol. 2002;20(4):1043–8.
de Jong AE, van Puijenbroek M, Hendriks Y, Tops C, Wijnen J, Ausems MG, Meijers-Heijboer H, Wagner A, van Os TA, Brocker-Vriends AH, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(3):972–80.
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18):1851–60.
Southey MC, Jenkins MA, Mead L, Whitty J, Trivett M, Tesoriero AA, Smith LD, Jennings K, Grubb G, Royce SG, et al. Use of molecular tumor characteristics to prioritize mismatch repair gene testing in early-onset colorectal cancer. J Clin Oncol. 2005;23(27):6524–32.
Salahshor S, Koelble K, Rubio C, Lindblom A. Microsatellite Instability and hMLH1 and hMSH2 expression analysis in familial and sporadic colorectal cancer. Lab Invest. 2001;81(4):535–41.
Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10(4):293–300.
Chang CL, Marra G, Chauhan DP, Ha HT, Chang DK, Ricciardiello L, Randolph A, Carethers JM, Boland CR. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol. 2002;283(1):C148–54.
Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, Giordano F, Johnson RS, Rockwell S, Glazer PM. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol. 2003;23(9):3265–73.
Bartley AN, Hamilton SR, Alsabeh R, Ambinder EP, Berman M, Collins E, Fitzgibbons PL, Gress DM, Nowak JA, Samowitz WS, et al. Template for reporting results of biomarker testing of specimens from patients with carcinoma of the colon and rectum. Arch Pathol Lab Med. 2014;138(2):166–70.
Dudley B, Brand RE, Thull D, Bahary N, Nikiforova MN, Pai RK. Germline MLH1 Mutations Are Frequently Identified in Lynch Syndrome Patients With Colorectal and Endometrial Carcinoma Demonstrating Isolated Loss of PMS2 Immunohistochemical Expression. Am J Surg Pathol. 2015;39(8):1114–20.
Rosty C, Clendenning M, Walsh MD, Eriksen SV, Southey MC, Winship IM, Macrae FA, Boussioutas A, Poplawski NK, Parry S, et al. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open. 2016;6(2), e010293.
Hagen CE, Lefferts J, Hornick JL, Srivastava A. “Null pattern” of immunoreactivity in a Lynch syndrome-associated colon cancer due to germline MSH2 mutation and somatic MLH1 hypermethylation. Am J Surg Pathol. 2011;35(12):1902–5.
Shia J, Zhang L, Shike M, Guo M, Stadler Z, Xiong X, Tang LH, Vakiani E, Katabi N, Wang H, et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol. 2013;26(1):131–8.
Roth RM, Haraldsdottir S, Hampel H, Arnold CA, Frankel WL. Discordant Mismatch Repair Protein Immunoreactivity in Lynch Syndrome-Associated Neoplasms: A Recommendation for Screening Synchronous/Metachronous Neoplasms. Am J Clin Pathol. 2016;146(1):50–6.
Walsh MD, Buchanan DD, Pearson SA, Clendenning M, Jenkins MA, Win AK, Walters RJ, Spring KJ, Nagler B, Pavluk E, et al. Immunohistochemical testing of conventional adenomas for loss of expression of mismatch repair proteins in Lynch syndrome mutation carriers: a case series from the Australasian site of the colon cancer family registry. Mod Pathol. 2012;25(5):722–30.
Yurgelun MB, Goel A, Hornick JL, Sen A, Turgeon DK, Ruffin MT, Marcon NE, Baron JA, Bresalier RS, Syngal S, et al. Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer Prev Res (Phila). 2012;5(4):574–82.
Bao F, Panarelli NC, Rennert H, Sherr DL, Yantiss RK. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am J Surg Pathol. 2010;34(12):1798–804.
Radu OM, Nikiforova MN, Farkas LM, Krasinskas AM. Challenging cases encountered in colorectal cancer screening for Lynch syndrome reveal novel findings: nucleolar MSH6 staining and impact of prior chemoradiation therapy. Hum Pathol. 2011;42(9):1247–58.
Vilkin A, Halpern M, Morgenstern S, Brazovski E, Gingold-Belfer R, Boltin D, Purim O, Kundel Y, Welinsky S, Brenner B, et al. How reliable is immunohistochemical staining for DNA mismatch repair proteins performed after neoadjuvant chemoradiation? Hum Pathol. 2014;45(10):2029–36.
Haraldsdottir S, Roth R, Pearlman R, Hampel H, Arnold CA, Frankel WL. Mismatch repair deficiency concordance between primary colorectal cancer and corresponding metastasis. Fam Cancer. 2016;15(2):253–60.
Bae JM, Kim JH, Kang GH. Molecular Subtypes of Colorectal Cancer and Their Clinicopathologic Features, With an Emphasis on the Serrated Neoplasia Pathway. Arch Pathol Lab Med. 2016;140(5):406–12.
Acknowledgements
We thank Shawn Scully for his work with image production and processing.
Funding
None.
Availability of data and materials
Not applicable.
Authors’ contributions
WC performed literature search and review, and constructed the manuscript. BS contributed to writing portion of the introduction and main text. WF designed the screening algorithm, provided the example cases, and critically read and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
None.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, W., Swanson, B.J. & Frankel, W.L. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 12, 24 (2017). https://doi.org/10.1186/s13000-017-0613-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-017-0613-8